Breaking Through the “Irreversible” Petrification of the Heart – ATTR-CM Treatment Shifts from Defense to Counterattack
April 10, 2026
Source: drugdu
 149
149
The story of transthyretin amyloid cardiomyopathy (ATTR-CM) begins with the structural instability of a protein. Under normal circumstances, TTR proteins transport thyroxine and vitamin A in the blood as tetramers, maintaining a stable structure and each fulfilling its function. However, under the influence of aging or specific gene mutations, the tetramer dissociates into monomers. These monomers misfold and aggregate into non-degradable amyloid fibers. Once these fibers deposit in the myocardial interstitium, the heart wall thickens and hardens, leading to decreased compliance, gradual loss of diastolic function, and ultimately heart failure.
For a long time, the clinical approach to these diseases was essentially defensive, using drugs to stabilize the protein structure, slow down the deposition rate, and prolong the disease course as much as possible. As for the fibers already embedded in the myocardium, it was generally accepted that there was nothing that could be done. However, in recent years, this situation has begun to change. Monoclonal antibody scavengers have provided early evidence in clinical trials of reversing myocardial damage, while gene editing technology aims to address the problem at the genomic level in one go. The underlying logic of treatment is shifting from "delaying deterioration" to "actively clearing" the disease.
This article traces the technological evolution of three generations of treatment strategies, outlining their respective clinical data, industry landscape, and unanswered questions.
Pathogenic mechanism: Tetramer disintegration and fibrillary deposition
ATTR-CM was previously classified as a rare disease. However, with advancements in screening methods in recent years, this assessment is being revised. The detection rate of ATTR-CM in patients with heart failure with preserved ejection fraction (HFpEF) is approximately 20%. In other words, a significant number of patients may have been broadly diagnosed with "diastolic heart failure" for a long time, without the true cause being identified.
The pathogenesis itself is not difficult to understand: TTR tetramers dissociate into monomers driven by aging or mutation. These monomers misfold, and the abnormally folded monomers aggregate into amyloid fibers. These fibers continuously accumulate in the intermyocardial spaces, causing the ventricular walls to thicken and harden, leading to a progressive loss of diastolic function. Theoretically, each link in this pathway could be a target for drug intervention. Which pathways are actually "completed," and where the "blockages" lie, will be analyzed in detail below.
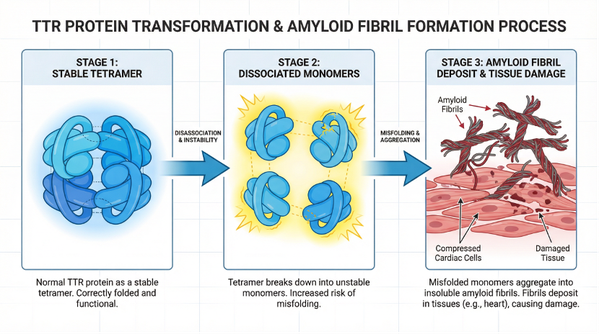 Figure 1: The process by which TTR protein dissociates from a stable tetramer into monomers, eventually misfolds and aggregates into amyloid filaments, leading to compression of cardiomyocytes.
Figure 1: The process by which TTR protein dissociates from a stable tetramer into monomers, eventually misfolds and aggregates into amyloid filaments, leading to compression of cardiomyocytes.
Data source: Compilation of publicly available data
An early signal that is easily missed
Clinical studies have revealed an interesting phenomenon: many ATTR-CM patients experience carpal tunnel syndrome or biceps tendon rupture 5 to 10 years before developing cardiac symptoms. The reason is simple—abnormal proteins tend to accumulate first in tissues under high mechanical stress, with ligaments and tendons being the first to be affected. The clinical value of this finding lies in providing a screening window. The problem is that carpal tunnel syndrome falls under orthopedics, while cardiomyopathy falls under cardiology, and information between the two departments is not shared in most hospitals. Establishing a referral awareness for high-risk groups within orthopedic departments would effectively move the diagnostic window forward by five to ten years.
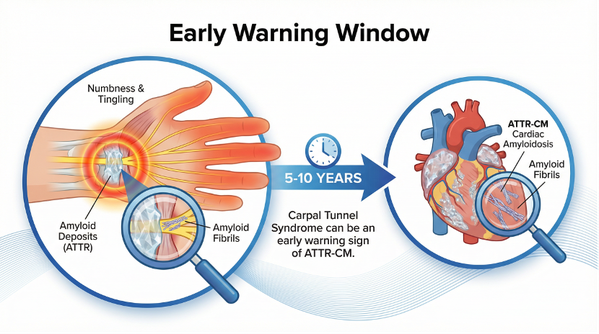 Figure 2: Timeline and pathological mechanism of carpal tunnel syndrome as an early warning signal of ATTR-CM
Figure 2: Timeline and pathological mechanism of carpal tunnel syndrome as an early warning signal of ATTR-CM
Data source: Compilation of publicly available data
Third-generation treatment strategy
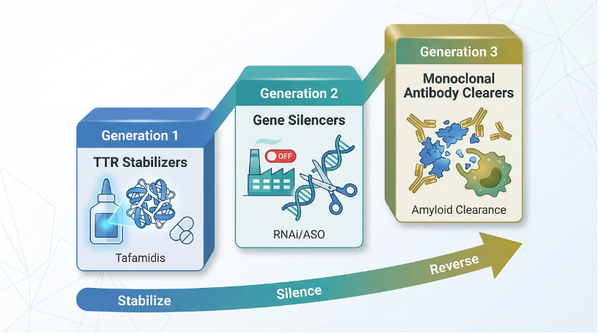 Figure 3: The three-generation evolution of ATTR-CM treatment regimens from stabilizers, gene silencing to monoclonal antibody clearance.
Figure 3: The three-generation evolution of ATTR-CM treatment regimens from stabilizers, gene silencing to monoclonal antibody clearance.
Data source: Compilation of publicly available data
Drug development for ATTR-CM proceeds sequentially along three directions. The table below provides a quick comparison:
First-generation defense: TTR stabilizer
The logic of stabilizers is straightforward: since the problem begins with the dissociation of tetramers, then use small molecules to lock in the tetramers and prevent them from falling apart.
Tafamidis is the pioneering drug in this class and the world's first approved disease-modifying drug for ATTR-CM. Clinical data shows a TTR stabilization efficiency of approximately 70%–80%, with a 36% reduction in all-cause mortality compared to placebo in the treatment group. Pfizer acquired the drug through its 2010 acquisition of FoldRx, and the Vyndaqel series is now a mainstay of its rare disease division: global sales of $5.45 billion in 2024, a year-on-year increase of 65%; and $1.62 billion in Q2 2025 alone. Acoramidis, developed by Eidos Therapeutics, is a later entrant. It has a stronger binding affinity and, mechanistically, mimics the T119M protective mutation to enhance hydrogen bond and salt bridge interactions, achieving a TTR stabilization rate close to 100%.
But whether it's Tafamidis or Acoramidis, the ceiling is the same: they only address the issue of "incremental" growth—preventing the formation of new fibers. As for existing fibers already embedded in the myocardial interstitium, the stabilizers "cannot reach" them.
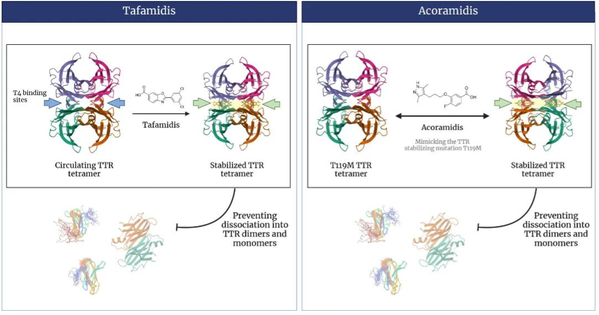 Figure 4: Mechanism of action of stabilizer drugs Tafamidis and Acoramidis
Figure 4: Mechanism of action of stabilizer drugs Tafamidis and Acoramidis
Image source: Medlive Cardiovascular
Second generation: TTR silencer shuts down the production line at the source.
Stabilizers lock in existing tetramers, but the liver continues to synthesize new TTR proteins. The idea of silencing agents is more radical: since downstream processes can't be controlled, shut down the production line upstream. TTR is almost entirely synthesized by the liver and encoded by a single gene, making it naturally suitable for targeting with siRNA and ASO technologies—the target is well-defined, concentrated in specific organs, and the delivery pathway is well-established.
Alnylam currently dominates this market segment. Patisiran (Onpattro) was the world's first approved siRNA drug (in 2018, for ATTR polyneuropathy), but it did not receive approval for ATTR-CM. Its successor product, Vutrisiran (Amvuttra), truly made a name for itself in the cardiomyopathy field. Using GalNAc conjugation technology, it is administered subcutaneously every three months, reducing TTR protein levels by approximately 88%. It is currently the only approved silencing agent for ATTR-CM. Its growth since launch has been rapid: sales reached $425 million in the first half of 2024, an 82% year-on-year increase.
The limitations of silencing agents and stabilizers are essentially the same: they turn off the tap, but the water already on the floor won't evaporate on its own. Therefore, for patients in later stages of the disease, the burden on existing myocardial fibers is the core issue, and simply blocking new growth is far from sufficient.
Third generation: TTR remover reaches existing fibers.
The first two generations of drugs share a common limitation: whether they lock in tetramers or shut down TTR synthesis, they only act on the "incremental" components. They are ineffective against existing fibers already embedded in the myocardial interstitium. The third-generation scavengers aim to solve precisely this problem.
The key technical aspect lies in targeting precision. In its normal tetrameric state, the TTR protein contains a tightly packed amino acid sequence that is unrecognizable from the surface. However, when the tetramer dissociates and the monomers misfold, this sequence is exposed, forming a so-called "cryptic epitope." The scavenging antibody specifically recognizes this marker, which only appears in the abnormal conformation—meaning that normally functioning TTR tetramers in circulating blood will not be mistakenly damaged. After binding to deposited fibrils, the antibody recruits macrophages to the deposition site via its Fc region, where macrophages then phagocytose and degrade the fibrils.
The two most advanced molecules currently are Cliramitug (NI006) from AstraZeneca/Alexion (originally developed by Neurimmune) and Coramitug (NNC6019) from Novo Nordisk (from Prothena). Both have entered Phase III clinical trials, and specific data will be discussed in the industry landscape section.
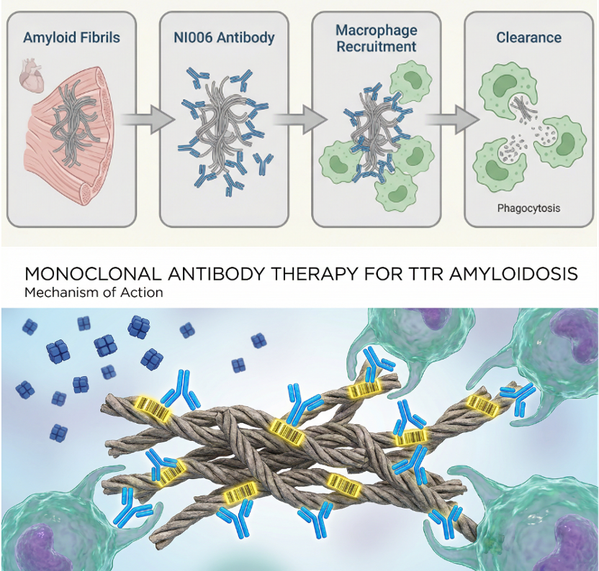 Figure 5: Monoclonal antibodies precisely target specific sequences on amyloid fibrils, guiding macrophages to clear them without affecting normal TTR tetramers.
Figure 5: Monoclonal antibodies precisely target specific sequences on amyloid fibrils, guiding macrophages to clear them without affecting normal TTR tetramers.
Data source: Compilation of publicly available data
Safe Dosage Design: Why We Can't Simply Increase Dosage Rate
There's an unavoidable safety issue with scavengers. The recruitment of large numbers of macrophages to the myocardial deposition sites essentially creates localized inflammation in an already dysfunctional heart. If the inflammatory response gets out of control, the consequences can be fatal for these patients. Therefore, the conventional approach of "starting with a low dose and gradually increasing" is very risky in this scenario: the process of increasing the dose is inherently risky.
Cliramitug's team opted for a more cautious approach: establishing a cross-species mechanistic pharmacokinetic/pharmacodynamic (PK/PD) model before proceeding to human trials. Specifically, they integrated rat pharmacokinetic data with in vitro results from human cardiac biopsies, computationally extrapolating the dosage regimen. The model yielded key parameters: an initial dose of 0.3 mg/kg, target occupancy controlled below 18%, an effective dose window of 10–60 mg/kg, and a predicted significant reduction in myocardial amyloid deposition within 4 to 10 months.
Subsequent clinical data confirmed these predictions. This "model first, then introduce" strategy may become a methodological template in the field of cleansing agents, where the safety window is extremely narrow.
Gene editing: The possibility of a single dose
Silencers require continuous administration to maintain low TTR levels: Amvuttra once every three months, Eplontersen once a month. Gene editing aims for something more radical: directly knocking out or modifying the TTR gene at the genome level, providing long-term benefits with a single dose.
Gene editing is particularly valuable for patients with advanced disease. While the progression of the disease slows after using stabilizers, their cardiac function continues to decline. If gene editing could permanently shut down the source of TTR (transient receptor agonist), combined with scavengers to treat existing deposits, this "source cut-off + reservoir clearing" combination is theoretically the most complete intervention. Of course, there are still two hurdles between theory and practice: long-term safety and off-target effects, neither of which have definitive answers yet.
Industry Landscape: Positioning on Three Tracks
The stabilizer market is currently Pfizer's territory. Tafamidis (Vyndaqel/Vyndamax) achieved global sales of $5.45 billion in 2024, a year-on-year increase of 65%. However, with its US patent expiring in 2028, the pressure from generic drugs is already mounting.
The silencer market is centered around Alnylam. Amvuttra saw rapid growth after its approval, capturing 35% of the new drug market share within three quarters of its launch. Eplontersen (Ionis/AstraZeneca) is currently in Phase III trials.
The landscape of the clearer drug race is the most intriguing. Novo Nordisk acquired Coramitug from Prothena, with potential milestone payments totaling $1.2 billion, and its Phase III CLEOPATTRA trial is progressing (approximately 1280 participants, expected to complete in 2029). Meanwhile, AstraZeneca/Alexion's Phase III DepleTTR-CM trial of Cliramitug is scheduled to begin in January 2024, with plans to enroll 1000 to 1181 participants, and is expected to complete in 2027-2028. Both companies are making significant investments in the same field almost simultaneously; whoever releases key data first will define the next generation of treatment standards.
With the widespread adoption of non-invasive diagnostic methods such as radionuclide imaging, the number of identified ATTR-CM patients continues to expand. The global ATTR amyloidosis treatment market is projected to reach US$14.28 billion by 2031.
In cardiovascular drug development, the first lesson is learning to respect the fragility of myocardial tissue. Myocardial tissue is one of the hard constraints in drug design. It is extremely sensitive to toxicity and has almost zero regenerative capacity. No matter how high the affinity of a molecule is, if there is a toxic signal in the hERG channel, the project basically cannot proceed. The history of ATTR-CM treatment is, in a sense, three tests of this lesson. The first time, we learned to use small molecules to glue back together proteins that were falling apart, preventing new damage from occurring. The second time, we directly shut down the line that produces faulty proteins in the liver. The third time, we tried to use antibodies to carry macrophages to clean up the fibers already embedded in the myocardium. Standing at the starting point now, we can look back and realize that ten years ago, the industry did not even think that the third step was possible.
But between scientific "possibility" and clinical "accessibility" lies a price list. Tafamidis' annual procurement cost in the US is approximately $268,000. Vutrisiran and Eplontersen exceed $500,000, and these are just single-drug formulations. If the optimal future approach involves a silencer plus a scavenger, or even a further gene editing, what would the total cost be? No one is currently willing to calculate that. Drug developers are accustomed to discussing molecular affinity, selectivity, and pharmacokinetic parameters. However, whether a drug ultimately benefits the public often doesn't directly depend on the Kd value, but rather on numerous objective factors related to the patient. This is largely beyond the scope of teaching theory, but it's a problem every new drug developer will face.
https://news.yaozh.com/archive/47737.html
By editor
Copyright©2026 Ddu. All rights reserved.
Read more on
- Chinese innovative drugs score a Blockbuster Deal! AstraZeneca buys out Suvolotinib for $1.5 billion July 15, 2026
- 【Company Recommendation】Shanghai Wantewant Technology Co., Ltd July 10, 2026
- Ascletis Pharma submits Investigational New Drug (IND) applications to the FDA for two new obesity drugs July 9, 2026
- Fengtou News | Highlight Pharmaceuticals’ Ganoxitinib Receives Breakthrough Therapy Designation from CDE July 9, 2026
- 【Company Recommendation】Emeishan Hongsen Biopharmaceutical Co., Ltd. June 26, 2026
your submission has already been received.
OK
Subscribe
Please enter a valid Email address!
Submit
The most relevant industry news & insight will be sent to you every two weeks.


