Optimization strategy for production process of RNA targeted drugs
October 3, 2024
Source: drugdu
 669
669
RNA targeted drugs are a completely new category of drugs that are completely different from small molecule drugs and antibody drugs. On the one hand, they can target mRNA, ncRNA, and other intracellular proteins through gene silencing to inhibit their expression and achieve the goal of treating diseases; On the other hand, new generation vaccines and protein replacement therapies can also be developed based on mRNA. RNA targeted drugs are undoubtedly a strategic frontier in biopharmaceutical innovation, with broad potential application scenarios in the future. The production process technology of mRNA industrialization is particularly important for the widespread application and industrialization of mRNA. Based on this, this article will focus on the optimization strategies for the production process of RNA targeted drugs.
Stability chemical modification enhances drug safety and efficacy
For RNA targeted drugs, chemical modification (excluding tissue targeting ligands) mainly has two basic functions: firstly, chemical modification can significantly improve the safety of drugs by weakening the immune response of endogenous immune sensors to dsRNA in cells; Secondly, by enhancing the ability of RNA drugs to resist degradation by endogenous and exogenous endonucleases, drug efficacy can be significantly improved;
For siRNA drugs, chemical modification can also enhance the selectivity of their antisense chains towards RISC loading, improve sequence selectivity to reduce off target RNAi activity, and alter physical and chemical properties to enhance delivery capability.
Specific chemical modifications can improve drug safety, metabolic stability, targeting, binding affinity, and silencing effect, greatly expanding the therapeutic window of drugs. Therefore, the absolute necessity of chemical modifications has been fully demonstrated in the early setbacks of clinical development of RNA targeted drugs. So far, all RNA targeted drugs approved by the FDA are chemically engineered RNA analogues that support the efficacy of chemical modifications.
Single stranded oligonucleotides targeting specific chemical modification categories only differ in sequence, but share similar physicochemical properties and therefore share common pharmacokinetic and biological characteristics. However, each chemical category is different, and even minor modifications between 2 '- methoxyethyl (2' - MOE) and 2 '- methoxy (2' - OMe) can lead to significant changes in efficacy and pharmacokinetics. Therefore, it is essential to accurately define the chemical properties of RNA targeted drugs.
The following figure shows the main chemical modification strategies under development, including nucleic acid backbone substitution, 2 'ribose modification, and 5' base modification, as well as other optional strategies. Currently, these methods are widely used to enhance the pharmacological properties of oligonucleotide drugs, thereby improving their pharmacokinetics, pharmacodynamics, and biological distribution.
In terms of the function of chemical modification, widely used modifications include 2 '- terminal ribose modification (such as using 2' - MOE, 2 '- OMe, 2' - F, etc. to replace the 2 '- terminal hydroxyl group), which can reduce immunogenicity, increase base pairing dissolution temperature (Tm), and enhance resistance to nucleases; Modifying sugar groups can greatly increase (lock nucleic acid LNA) or decrease (unlock nucleic acid UNA) the binding affinity with RNA; Changes in the nucleic acid backbone can increase non-specific protein binding, eliminate backbone charges, and increase hydrophobicity (morpholino PMO or peptide nucleic acid DNA) to resist nuclease degradation.
Chemical modification strategies for RNA targeted drugs
Data source: PubMed
The specific types of chemical modifications and their effects are as follows:
2 'ribose substitution strategy
The hydroxyl group (- OH) of oligonucleotides at the 2 'end of ribose can be replaced by substituents such as MOE, OMe, F, etc., which are used to reduce immunogenicity, increase resistance to nucleases, improve plasma stability, and thus prolong drug action.
(1) 2 '- Methoxyethyl (2' - MOE): can enhance drug PK, prolong clearance half-life to 2-4 weeks, improve binding ability and efficacy with targeted mRNA, and reduce cytotoxicity;
(2) 2 '- Methoxy (2' - OMe): can improve drug PK and stability, moderately enhance efficacy and reduce immunogenicity;
(3) 2 '- Fluorine (2' - F): It can enhance the binding ability between drugs and targeted mRNA, but cannot improve stability and PK. It is more suitable for siRNA drugs with RISC mechanism. Its modified nucleotide metabolites have the possibility of integrating into host cell DNA or RNA, which can lead to partial degradation of nuclear proteins.
In practical applications, modifications at the 2 'end of ribose are incompatible with RNase H activity, which means they are typically used for spatially hindered ASO or for flanking sequences of Gapmer ASO (such as Miumersen and Inotersen introducing 2' - MOE modifications in flanking sequences).
In addition, 2 '- MOE modification is usually not included in siRNA design, and the structural requirements of Ago2 limit the types of chemical modifications that can be used. 2' - MOE has been shown to be very useful for ASO with RNase H activity, but does not support siRNA binding to Ago2.
5 'end nucleobase modification strategy
5 '- Methylcytidine and 5' - Methyluridine modifications can enhance the binding ability of drugs to target mRNA and reduce immunogenicity, but are only used for heterocyclic modifications. Pyrimidine methylation can increase the melting temperature of each modified oligonucleotide by about 0.5 ℃, enhance the binding ability and stability of drugs with target mRNA, and is commonly used in ASO drugs (such as those being developed by Ionis Pharmaceuticals).
Riboskeleton modification strategy: The phosphodiester bond of oligonucleotides can be replaced by a thiophosphate (PS) bond, which means that one non bridging oxygen atom of the phosphate group between nucleotides is replaced by sulfur. It has been widely used in the development of RNA targeted drugs. PS riboskeleton is a highly effective modification that has a dual effect of nuclease resistance and promoting binding with plasma proteins, thereby reducing renal clearance, increasing drug circulation time, improving drug pharmacokinetics, and increasing drug clearance half-life to 1-3 days.
The PS riboskeleton supports RNA targeted drugs with multiple mechanisms of action, especially in the application of Gapmer ASO and GalNAc siRNAs with high efficiency. PS riboskeleton modification is easy to tolerate in ASO design and does not disrupt RNase H activity. It is used in most marketed ASO drugs and can be absorbed by most cells after systemic administration without the need for targeted ligands.
In contrast, siRNA containing PS modification at each linkage site has lower activity than equivalent phosphodiester (PO) siRNA, so siRNA containing PS modification is usually only modified at the end (such as Patisran, which has been approved for marketing).
One disadvantage of adding PS modifications in RNA targeted drugs is that each PS modification introduces a stereocenter with two possible chiral orientations, so oligonucleotides with n PS modifications are a mixture of 2n racemates.
These two orientations have significantly different pharmacokinetic and pharmacodynamic properties. Although the PS bond of Sp orientation has better resistance to nuclease cleavage, compared with Rp orientation, they also tend to reduce the dissolution temperature of the side bases of the base and lower stability.
Due to the heterogeneity of molecules, which is often detrimental to their clinical development, future RNA targeted drugs may benefit from recently developed PS modified oligonucleotide stereoselective synthesis techniques, such as Wave Life Sciences, which has developed a scalable method to synthesize oligonucleotides with fixed stereochemistry on each PS strand and is developing oligonucleotide drugs with fixed stereochemistry for various indications.
Another disadvantage of PS modification is that they reduce the binding affinity of oligonucleotides to their targets, which can be compensated for by combining other types of modifications.
It is worth noting that PS modification enhances drug resistance to cellular nucleases, which can lead to drug tissue retention and long-lasting drug effects. To cope with adverse reactions such as toxicity caused by prolonged gene silencing, the binding of one or more PO bonds can regulate the durability of oligonucleotides by reducing the stability of their nucleases.
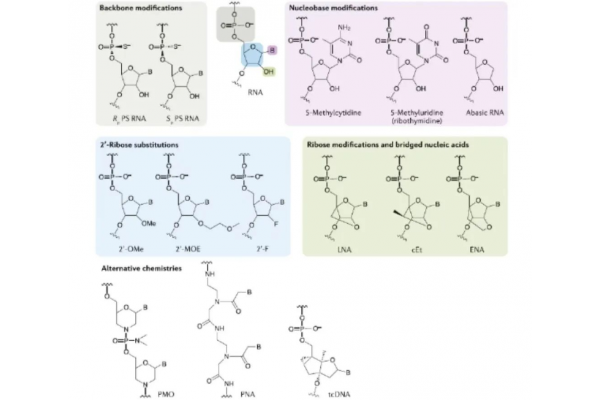
2 'ribose modification and nucleotide bridging strategy
Nucleotide bridging (BNAs) is confined to a fixed conformation by bridging between the second and fourth carbon atoms of nucleotides. The most commonly used bridging strategies are LNA, cEt, and ENA. BNAs enhance drug resistance to nucleases and affinity for target mRNA (in LNA, the melting temperature of each modified nucleotide increases by 3-8 ℃).
Although BNA modified nucleotides are not compatible with RNase h, BNA modification can be incorporated into the flanking region of Gapmer ASO or used in spatially hindered ASOs to improve target binding. It is worth noting that LNA modified RNA targeted drugs have been observed to have hepatotoxicity and nephrotoxicity in some clinical trials, increasing sequence related risks. Further attention is needed in the future.
Other modification strategies: Glyphosate oligonucleotides (PMO) are a powerful class of synthetic oligonucleotide analogs, belonging to the third generation of antisense oligonucleotides in the history of nucleic acid drug development. The electrically neutral morpholine structure of PMO gives it high binding affinity and strong enzymatic stability. So far, two PMO modified drugs (Eteplirsen and Golodirsen) have been approved for market by the FDA.
PMO has the characteristics of high stability and safety. Due to its neutrality at physiological pH, it does not support RNase H1 activity, so it is mainly used for drugs with steric hindrance mechanism. One disadvantage of PMO modification is its lack of ability to bind to albumin, resulting in lower PK characteristics. This means that it can be quickly cleared through renal excretion, leading to lower efficacy, and therefore requires an increase in drug dosage.
It is worth noting that the PMO main chain contains chiral centers, which means that PMO drugs must be a mixture of racemic compounds. Unlike the PS modification mentioned above, the impact of defining PMO stereochemistry has not been studied so far.
Chemical modification types and efficacy of RNA targeted drugs
Data source: PubMed
The delivery system effectively enhances targeting and bioavailability
Regardless of chemical modifications, the size, hydrophilicity, and charge of RNA targeted drugs pose additional challenges to drug development in terms of systemic circulation, tissue extravasation, cellular uptake, and endosome escape, as nucleic acid drugs need to enter the cell and complete endosome escape in order to exert pharmacological effects.
In order to overcome the obstacles of low cellular uptake and intracellular escape efficiency of nucleic acid drugs, delivery systems are necessary to enhance drug targeting and bioavailability.
At present, delivery systems developed for RNA targeted drugs mainly include lipid nanoparticles (LNPs), polymers, nucleic acid nanostructures, exosomes, etc. RNA targeted drugs can also covalently bind to specific ligands, ranging from relatively small molecules (such as aptamers, GalNAc, etc.) to large molecules (such as peptides, antibodies, etc. Bioconjugation). Ligand directed delivery is expected to improve targeting for specific types of cells.
In addition, delivery systems are currently the main bottleneck restricting the development of RNA targeted drugs worldwide. Lipid nanoparticles (LNPs) and N-acetylgalactosamine (GalNAc) have been validated in approved nucleic acid drugs, and the development of other delivery systems is also being validated in clinical practice.
Lipid nanoparticles (LNPs) were initially developed as delivery systems for siRNA drugs in vivo. LNPs are complex structures (~100 nm) and have also been used to deliver large RNA molecules such as mRNA in vivo. LNPs can encapsulate a large amount of RNA, protecting it from RNase degradation and renal clearance.
It has been clinically proven that the addition of polyethylene glycol (PEG) lipids can enhance their circulation time in the body, as it forms a spatial barrier around the LNP surface and protects LNPs from interacting with plasma proteins, which will target LNPs for degradation by MPS. Therefore, the use of PEG lipids in LNPs has become standard.
However, the use of PEG lipids is a double-edged sword, as the same spatial barrier also inhibits the interaction between LNPs and the cell membrane, as well as the efficiency of subsequent escape from the endosome. Therefore, fine-tuning the number of PEG lipids and the length of PEG chains is crucial.
The disadvantage of clinical application of LNPs is that their delivery is mainly limited to the liver and reticuloendothelial system, as the sinusoidal capillary epithelium of this tissue provides sufficient space to allow these relatively large nanoparticles to enter. In addition, local delivery of LNPs has been successfully used to deliver siRNA to the central nervous system.
LNPs can be further functionalized by peptides, PEG, or other ligands that provide cell specific targeting. However, it is worth noting that the increase in complexity of LNPs can complicate their manufacturing and potentially increase their toxicity, which is a major issue that may limit their clinical application.
For example, siRNA drugs wrapped in LNPs (such as Patisiran) require pre-treatment with steroids and antihistamines before intravenous injection to eliminate unnecessary allergic reactions. In addition, biodegradable ionized lipids may enter the preclinical development stage in the next 2-5 years, which is expected to significantly increase the tolerated dose of drugs.
Application of LNPs delivery system in siRNA drugs
Data source: Nature
In addition to delivery systems such as LNPs, RNA targeted drugs can also covalently bind to specific ligands, ranging from relatively small molecules (such as aptamers, GalNAc, etc.) to large molecules (such as peptides, antibodies, etc. Bioconjugation). Ligand directed delivery is expected to improve targeting of specific cell types.
Among them, GalNAc is the most widely used ligand for nucleic acid drugs targeting liver cells. Currently, about one-third of RNA targeted drugs in clinical trials bind to multivalent GalNAc ligands, targeting the sialic glycoprotein receptors (ASGPRs) on the surface of liver cells, greatly improving the targeting and bioavailability of nucleic acid drugs.
High levels of trimeric ASGPRs (approximately 105-106 per cell) are expressed on the surface of liver parenchymal cells. ASGPRs can specifically bind to GalNAc at neutral pH and release GalNAc in acidic environments (pH 5-6). Subsequently, the released ASGPRs can be recovered to the cell surface for reuse.
Therefore, the suitable physiological conditions of the liver, the unique characteristics of ASGPRs, the non-toxic nature of GalNAc ligands, and the ease of coupling make it a nearly ideal method for delivering systemic nucleic acid drugs to liver cells.
In addition, GalNAc bound oligonucleotides can be efficiently administered through SC (adipose tissue below the epidermis and dermis) injection. Subcutaneous injection of drugs releases into the systemic circulation at a slower rate and can also enter the lymphatic system, giving cell receptors more time to regulate absorption. At the same time, subcutaneous injection is faster and easier, reducing the burden of treatment.
More importantly, currently in clinical practice, the incidence of local adverse events related to subcutaneous administration of GalNAc bound oligonucleotide drugs is relatively low, and the additional safety and tolerability will significantly support less weekly administration, which is a comparative advantage over LNPs delivery systems.
The end of siRNA drugs can potentially connect to conjugates, but try to avoid binding to the 5 'end of the guide chain as much as possible, because the phosphate at this end specifically contacts the Ago2 central region side chain residue required for RNAi activity. Coupling to the guest chain is usually done to not hinder the targeted silencing activity of the guide chain, and correspondingly reduce the off target gene silencing potential of the guest chain.
Conjugated linkers can be designed to automatically decompose upon cell entry, or can be achieved through the use of acid resistant linkers cleaved in the endosome, reduced disulfide linkers in the cytoplasm, or Dicer substrate siRNA designs.
Various other delivery systems are also being developed in clinical practice:
(1) Adapter body
The coupling of therapeutic oligonucleotides with nucleic acid aptamers has also been used to enhance targeted delivery of siRNA and ASO. Aptamers can be considered as chemical antibodies with high affinity for their respective target proteins, but they have many advantages compared to antibodies, such as simple production, low cost (i.e. through chemical synthesis), small size, and lower immunogenicity.
(2) Polypeptides
Peptides are an attractive ligand source that can endow therapeutic oligonucleotide conjugates with tissue/cell targeting and cell penetration or endocytosis capabilities. CPP (also known as protein transduction domain) is typically short (usually about 30 amino acids), and one of its most promising applications is its direct chemical coupling to charged neutral ASO drugs such as PMO or PNA modifications.
(3) Endogenous exosomes
Extracellular vesicles are composed of lipid bilayer vesicles and are believed to be an endogenous delivery system that transmits information between cells through the transport of complex macromolecules (i.e. nucleic acids, proteins, and lipids), without activating the innate or acquired immune system.
Extracellular vesicles have multiple advantages in oligonucleotide drug delivery, such as high safety, the ability to cross biological membranes (including the blood-brain barrier), and the ability to increase systemic half-life by inhibiting MPS clearance and improving cellular uptake. The main challenge of using extracellular vesicles for nucleic acid drug delivery is effective loading, which can be achieved through methods such as electroporation, and other pathways are also being developed.
(4) DNA nanostructure
DNA nanostructures can be used for the delivery of oligonucleotides, mainly including DNA origami, polygonal DNA nanostructures, etc. DNA nanostructures used for nucleic acid delivery applications are usually modular, incorporating nucleic acid drugs in the design of the structure itself. Currently, corresponding ASO and siRNA drugs have been designed in clinical practice.
DNA nanostructures do not accumulate in the liver and can be designed as small nanostructures (about 20nm), which means that extrahepatic transmission is possible. At present, Jingze Biotechnology in China is developing a nucleic acid carrier platform based on DNA tetrahedral framework for the delivery of drugs such as oligonucleotides and small molecules.
In addition to delivery systems, the method and location of administration have a profound impact on the bioavailability and biodistribution of RNA targeted drugs. RNA targeted drugs in clinical development have been applied to GalNAc binding and LNP formulation ASO and siRNA drugs through local inhalation (in the lungs), local injection (in the eyes, heart, or cerebrospinal fluid), which avoids hepatic first channel metabolism.
At present, most of the candidate drugs developed in clinical practice are targeted at the liver. However, RNA targeted therapy is being developed to use local administration to avoid the limitations of systemic drug distribution, retaining RNA targeted drugs in local areas to prolong release time. This work is currently an active academic research field.
BERAs represent the direction of optimizing the production process of RNA targeted drugs
Pure homogeneous formulations are essential for the biological activity and therapeutic window of RNA targeted drugs in the human body.
At present, RNA targeted drugs are mainly produced through chemical synthesis or by relying on recombinant T7 RNA polymerase for in vitro transcription. However, the limitations of these produced RNA preparations are the addition of excessive artificial modifications to natural oligonucleotides or the lack of necessary post transcriptional modifications, which may lead to different folding characteristics, biological activity, and safety.
In addition, the cost of obtaining higher orders of siRNA or ASO materials through the above production methods is still expensive, and the length or size of synthesized oligonucleotides is also limited. Therefore, it is necessary to develop more economical, efficient, and scalable production methods.
The large-scale biological fermentation production technology (BERAs) is expected to provide a large number of biological RNA preparations with appropriate folding and natural modifications, which are crucial for the high-order structure, stability, activity, and safety of RNA.
In principle, the target RNA coding sequence is introduced into a plasmid, and the designed plasmid is transfected into host cells grown under appropriate conditions for transcription.
However, heterogeneous RNA is highly sensitive to intracellular nucleases, and the resulting RNA preparations may not accumulate to ideal levels. The use of stable RNA scaffolds (such as rRNA, tRNA), siRNA binding p19 binding proteins, or direct overexpression in RNase III deficient bacteria under these conditions prevents the target RNA preparations from being degraded by intracellular nucleases, which is an effective way to improve production efficiency.
RNA stability scaffolds used in BEARs production
Data source: Nature
Another important consideration in the production process is the complexity, uniformity, stability, and cytotoxicity of the delivery system and excipients, such as nanoparticles, polymers, peptides, etc.
Lipids and polymer nanoparticles have been widely used to improve the pharmacokinetic properties of RNA targeted drugs, but their production processes are complex and the finished products often have a certain degree of heterogeneous particle composition, making it more difficult to establish effective therapeutic windows.
In addition, nanoparticles become unstable during storage or after administration, releasing decomposition products that can lead to untraceable cytotoxicity. These issues need to be addressed to the greatest extent possible in the subsequent production process optimizati
By editor
Copyright©2026 Ddu. All rights reserved.
Read more on
- its stock price has shrunk by 90% from its issue price, and although it has finally received approval for its first product, the market landscape has already changed. August 4, 2026
- China’s first generic tiotropium bromide inhalation spray approved. August 4, 2026
- Ibrutinib tablets receiving drug registration certificate is considered equivalent to passing the consistency evaluation August 4, 2026
- Isaconazole Sulfate for Injection and Enzalutamide Tablets are expected to be selected in the 12th batch of national centralized procurement August 4, 2026
- Leads Biolabs receives IND approval from the National Medical Products Administration for the treatment of metastatic colorectal cancer. August 3, 2026
your submission has already been received.
OK
Subscribe
Please enter a valid Email address!
Submit
The most relevant industry news & insight will be sent to you every two weeks.


