Brief Bioinform | Li Shiliang/Li Honglin team develops AI method GR pKa value prediction for small molecules based on retention mechanism
September 27, 2024
Source: drugdu
 333
333
Zhiyao Bang
The following article is from DeDrug, authored by Li's Lab
On August 22, 2024, the team of Professor Li Shiliang/Li Honglin from East China University of Science and Technology/East China Normal University published a research paper titled "GR pKa: A message feeding neural network with retention mechanism for pKa prediction" in the journal Briefing in Bioinformatics.
 This paper introduces an innovative deep learning model GR pKa for predicting the pKa values of small chemical molecules. This model combines multiple fidelity learning, quantum chemistry features, and retention mechanisms to achieve rapid and accurate prediction of pKa values of small chemical molecules, providing a powerful new tool for research in the fields of chemistry and pharmacy.
This paper introduces an innovative deep learning model GR pKa for predicting the pKa values of small chemical molecules. This model combines multiple fidelity learning, quantum chemistry features, and retention mechanisms to achieve rapid and accurate prediction of pKa values of small chemical molecules, providing a powerful new tool for research in the fields of chemistry and pharmacy.
Research background
In the process of drug discovery and design, the acid-base dissociation constant (pKa) of molecules has a significant impact on the characteristics (absorption, distribution, metabolism, excretion, and toxicity) and biological activity of ADMET, and is therefore highly valued. However, traditional experimental methods for determining pKa values are both laborious and complex. At the same time, existing prediction methods have limitations in terms of the quantity and quality of training data, as well as their ability to handle complex molecular structures and physicochemical properties, which limits the predictive accuracy and generalization ability of the model. Therefore, developing a method that can quickly and accurately predict the pKa value of molecules will to some extent contribute to the structural modification of molecules, thereby assisting the process of new drug development.
In response to the above issues, the research team has developed an innovative pKa prediction method - GR pKa (Graph Retention pKa). This method is based on message passing neural networks and combines multiple fidelity learning strategies to achieve high-precision prediction of molecular pKa values. The GR pKa model specifically integrates five quantum mechanical (QM) properties related to molecular thermodynamics and dynamics as key features for characterizing molecules. Of particular note is that this model introduces the recently proposed retention mechanism into the message passing stage for the first time, which significantly improves the model's ability to capture and update molecular information. After testing on multiple datasets, the GR pKa model outperforms several currently leading models in predicting macroscopic pKa values. The prediction results on the SAMPL7 dataset showed that the model achieved high-level results with a mean absolute error (MAE) of 0.490, root mean square error (RMSE) of 0.588, and coefficient of determination (R2) of 0.937, verifying its efficiency and reliability in predicting pKa values.
Research method
GR pKa mainly consists of two processes, namely pre training and fine tuning, as shown in Figure 1a. This study constructed a low fidelity dataset based on the pKa values of small molecules calculated by Chemaxon, and pre trained the model using this dataset. By learning on this massive low fidelity dataset, the model is able to gain a deeper understanding of the quantitative structure-activity relationship between compound molecular structures and calculated pKa values. However, due to the significant difference between the calculated pKa values and the experimental pKa values, relying solely on this dataset for training and learning results in inaccurate predictions. Therefore, this study fine tuned the pre trained model to improve the accuracy of predictions. During the fine-tuning process, this study selected a small-scale high fidelity dataset consisting of experimental pKa values to further optimize the pre trained model. Through this adjustment, the model achieved a transition from calculated pKa values to experimental pKa values, significantly improving the accuracy of model predictions. Due to the high correlation between calculated pKa values and experimental pKa values, it is feasible to transfer between the two, and this transfer can achieve positive improvement in prediction results while effectively avoiding negative transfer.
The overall framework of the model is a message passing network, with the SMILES formula of molecules as input, as shown in Figure 1b. Firstly, this method constructs atomic feature matrices, bond feature matrices, and three interatomic matrices as local encodings for molecules. At the same time, quantum chemical properties with clear physical and chemical meanings were selected as the global characteristics of the molecule. In the processing of the key feature matrix, the model iteratively updates the feature matrix through the key retention module and key update function in the message passing layer. After the message passing phase is completed, feature integration is achieved by aggregating the hidden states of the incoming keys. Then, concatenate the atomic feature matrix with the multi head atomic preservation module to obtain the atomic representation. In the atomic reservation module, the distance matrix, adjacency matrix, and charge matrix, which are three scaled interatomic matrices, are added to the weights of each reservation head as bias terms. These matrices represent the distance, adjacency relationship, and charge information between atoms, respectively. Finally, the learned hidden states of atoms are aggregated into a molecular vector and concatenated with pre-defined quantum chemical features. This combination of features is then input into the fully connected layer for predicting molecular pKa values.
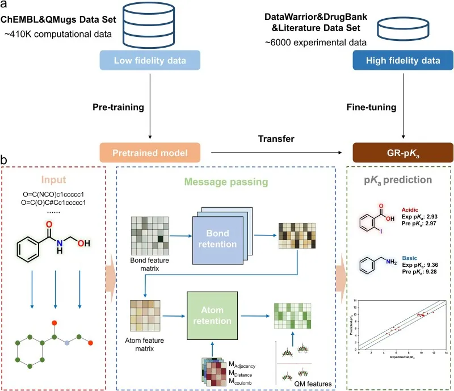 Figure 1 Overview of GR pKa method. a: Multi fidelity learning strategy for GR pKa. b: The Model Framework of GR pKa
Figure 1 Overview of GR pKa method. a: Multi fidelity learning strategy for GR pKa. b: The Model Framework of GR pKa
Research results
1. Method comparison
This study first compares the methods on the high-quality external dataset E-pKa dataset constructed. As shown in Figure 2, among all machine learning methods, XGBoost performs the best, and its prediction results are to some extent better than the deep learning method AttentiveFP. However, overall, the predictive performance of machine learning methods is weaker than that of MolGpKa and GR pKa models, indicating that deep learning models using molecular graph structures as inputs have significant superiority in predicting molecular pKa values. In addition, among all the compared prediction methods, the GR pKa model showed the best performance on both acidic and alkaline datasets. On the acidic dataset, GR pKa achieved the lowest MAE, RMSE, and highest R2, with values of 0.528, 0.758, and 0.939, respectively; On the alkaline dataset, the lowest MAE (0.447) and RMSE (0.651), as well as the highest R2 (0.897), were also achieved. These results fully demonstrate the superiority and accuracy of the GR pKa model in predicting pKa values.
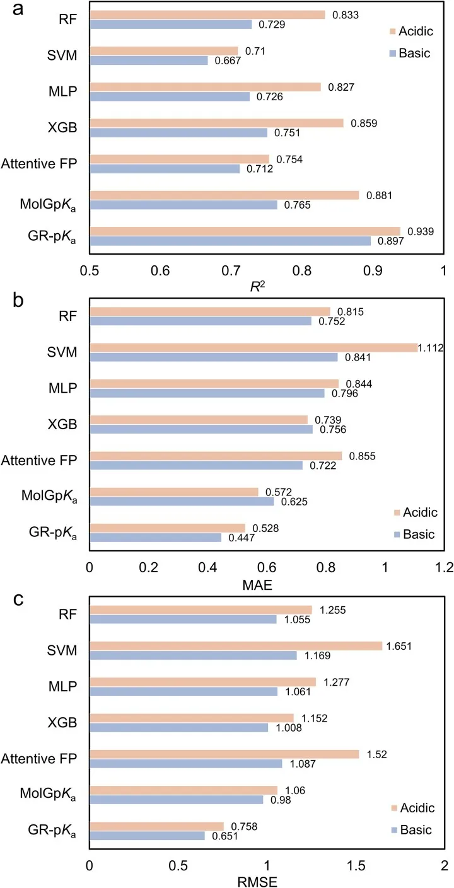 Figure 2 Performance of Method on E-pKa Dataset
Figure 2 Performance of Method on E-pKa Dataset
2. Ablation experiment
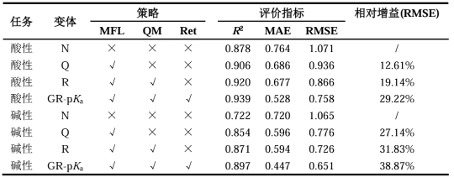
In order to further evaluate the effectiveness of the multi fidelity learning, QM features, and retention mechanisms used in this study, as well as their contribution to prediction accuracy, ablation experiments were designed and implemented for quantitative comparison. The results are shown in Table 1. The experimental results show that the use of multi fidelity learning effectively expands the predictive ability of the model, achieving effective fusion of low-quality data and high-quality data. Meanwhile, the introduction of a priori quantum chemical properties with clear physical and chemical meanings as auxiliary representations of molecular features significantly improves the accuracy of predicting molecular pKa values. In addition, researchers generally agree that the fewer chemical bonds between two atoms, the stronger their interactions, highlighting the importance of local information within the molecule. In this study, the application of retention mechanism in feature update learning can focus more on local information, which is consistent with basic chemical principles to some extent. Therefore, this study suggests that compared to attention mechanisms, retention mechanisms have certain advantages in capturing molecular information. The results of the ablation experiment further confirmed the hypothesis of this study: introducing a retention mechanism during the information transmission stage significantly improved the model's ability to update and learn chemical information.
Table 1 Experimental results of ablation of E-pKa model on external dataset
3. External verification
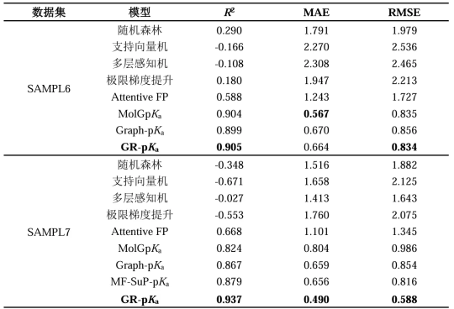
This study also conducted method testing and comparison on two recognized benchmark test sets SAMPL6 and SAMPL7, and the results are shown in Table 2. From the results in the table, it can be seen that all four machine learning methods perform poorly, while the other molecular graph based methods perform well. On the SAMPL6 dataset, both the GR pKa method and the MolGpKa method in this study demonstrated good performance; In the SAMPL7 dataset, the GR pKa method achieved a leading level in all three evaluation metrics and showed significant performance improvement compared to other methods. In addition, according to the detailed prediction results of the SAMPL7 test set in Figure 3, it can be seen that except for the prediction result of molecule SM31 exceeding the ± 1 region, all other molecules are within the region. This phenomenon indicates that our research method has significant advantages in accurately predicting molecular pKa values.
Table 2 Performance of the model on the SAMPL6 and SAMPL7 datasets
Summary
In this study, the research team successfully developed an innovative small molecule pKa value prediction method GR pKa based on a message passing neural network framework. In response to the problem of scarce experimental pKa value data, this study adopted a multi fidelity learning strategy, effectively integrating low-quality calculated pKa value data and high-quality experimental pKa value data to enhance the model's generalization ability and prediction accuracy. In addition, this study also introduced a priori quantum chemical properties with clear physical and chemical significance as auxiliary representations of molecular features, further improving the accuracy of predictions. Another innovation of this project is the first application of a retention mechanism to the message passing stage, optimizing feature update learning. Through system evaluation, the model demonstrated excellent performance on both an external dataset and two recognized benchmark datasets. In addition, through ablation experiments, this study clarified the contribution of different strategies to improving predictive performance and verified the effectiveness of the adopted strategies.
Miao Runyu from East China University of Science and Technology and Liu Danlin, a postdoctoral fellow from East China Normal University, are co first authors of this study. Mao Liyun, Chen Xingyu, Zhang Leihao, Yuan Zhen, and Shi Shanshan from East China University of Science and Technology also participated in the work. Professors Li Shiliang and Li Honglin from East China Normal University/East China University of Science and Technology are the corresponding authors of this article. This work has received funding support from the National Natural Science Foundation of China (821736908242510482150208) and the National Key Research and Development Program (2022YFC3400501, 2022YFC3400504).
By editor
Copyright©2026 Ddu. All rights reserved.
Read more on
- Chinese innovative drugs score a Blockbuster Deal! AstraZeneca buys out Suvolotinib for $1.5 billion July 15, 2026
- 【Company Recommendation】Shanghai Wantewant Technology Co., Ltd July 10, 2026
- Ascletis Pharma submits Investigational New Drug (IND) applications to the FDA for two new obesity drugs July 9, 2026
- Fengtou News | Highlight Pharmaceuticals’ Ganoxitinib Receives Breakthrough Therapy Designation from CDE July 9, 2026
- 【Company Recommendation】Emeishan Hongsen Biopharmaceutical Co., Ltd. June 26, 2026
your submission has already been received.
OK
Subscribe
Please enter a valid Email address!
Submit
The most relevant industry news & insight will be sent to you every two weeks.


